NOTE: If using in an HTML file, just simply include the <script src=””> tag with the appropriate location of the file
constDatabaseManager=require('./DatabaseManager');//constdbManager=newDatabaseManager('myDatabase',1,'myStore','id');dbManager.init().then(()=>{console.log('Database initialized and ready to use');}).catch(err=>{console.error('Error initializing the database:',err);});
2. Set a value in the store
dbManager.set('username','johnDoe').then(()=>{console.log('Value set successfully');// Get the value from the storereturndbManager.get('username');}).then(value=>{console.log('Retrieved value:',value);}).catch(err=>{console.error('Error setting or getting the value:',err);});
3. Delete a value in the store
dbManager.delete('username').then(()=>{console.log('Value deleted successfully');}).catch(err=>{console.error('Error deleting the value:',err);});
4. List all keys in the store
dbManager.list().then(keys=>{console.log('Keys in the store:',keys);}).catch(err=>{console.error('Error listing keys:',err);});
5. Retrieve all entries from the store
dbManager.getAll().then(entries=>{console.log('All entries:',entries);}).catch(err=>{console.error('Error retrieving all entries:',err);});
6. Clear all entries from the store
dbManager.clear().then(()=>{console.log('Store cleared successfully');}).catch(err=>{console.error('Error clearing the store:',err);});
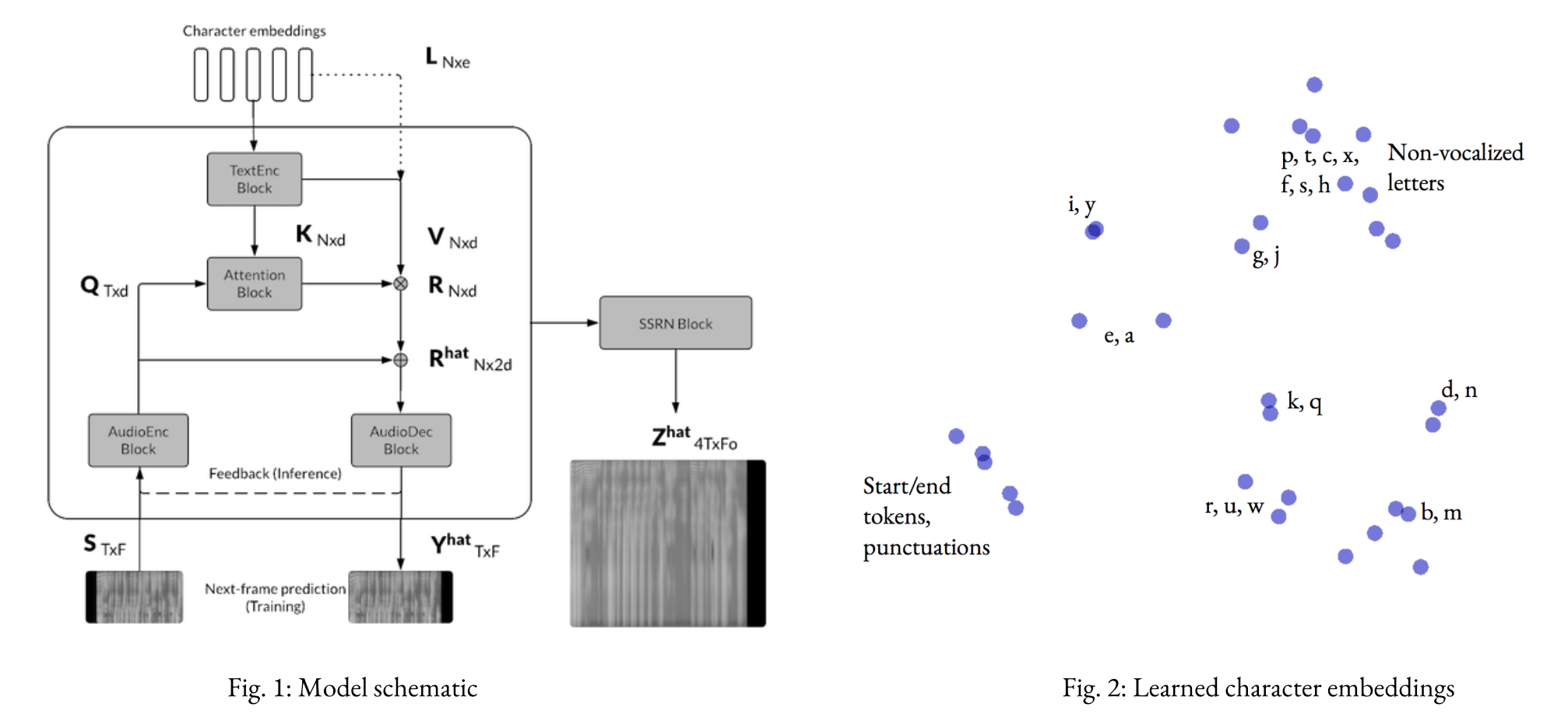
Attention, I’m Trying to Speak: End-to-end speech synthesis (CS224n ’18)
Implementation of a convolutional seq2seq-based text-to-speech model based on Tachibana et. al. (2017).
Given a sequence of characters, the model predicts a sequence of spectrogram frames in two stages (Text2Mel and SSRN).
As discussed in the report, we can get fairly decent audio quality with Text2Mel trained for 60k steps, SSRN for 100k steps. This corresponds to about (6+12) hours of training on a single Tesla K80 GPU on the LJ Speech Dataset.
- runs (contains checkpoints and params.json file for each different run. params.json specifies various hyperameters: see params-examples folder)
- run1/params.json ...
- src (implementation code package)
- sentences (contains test sentences in .txt files)
train.py
evaluate.py
synthesize.py
../data (directory containing data in format below)
- FOLDER
- train.csv, val.csv (files containing [wav_file_name|transcript|normalized_trascript] as in LJ-Speech dataset)
- wavs (folder containing corresponding .wav audio files)
Script files
Run each file with python <script_file>.py -h to see usage details.
/** * Transform mini-app asset urls. * @see https://github.com/NervJS/taro/blob/next/packages/taro-mini-runner/src/webpack/vue3.ts#L43-L50 */exportdeclareconsttransformMiniappAssetUrls/** * Transform H5 asset urls. * @see https://github.com/NervJS/taro/blob/next/packages/taro-webpack-runner/src/config/vue3.ts#L49-L62 */exportdeclareconsttransformH5AssetUrls/** * Declare native mini-app tags, so that miniapp native components * such as `picker`, `swiper`, `scroll-view` and etc. * will be treated as native tags, thus not to be resolved as components. */exportdeclarefunctionisMiniappNativeTag(tag: string): boolean;/** * Transform tags for h5 components. * For example, tag `view` will be transformed to `taro-view`, * so that it will be compiled to `resolveComponent('taro-view')`. */exportdeclarefunctiontransformH5Tags(): NodeTransform;/** * Transform `taro-env` or `taroEnv` prop, * and remove node that is not for the specified platform * @param platform `'mini' | 'h5'` */exportdeclarefunctiontransformEnv(platform?: 'mini'|'h5'): NodeTransform;/** * Transform `onClick` to `onTap` on native tags. */exportdeclareconsttransformClick: NodeTransform;
{
"jupyterCommand": "/usr/local/bin/jupyter-notebook", // executable path for Jupyter Notebook"jupyterPort": 8888, // server port"jupyterHome": "~", // root folder"openBrowserOnStartup": true, // set true if let Juno open browser after launch"preferLab": false// open Jupyter Lab instead of Jupyter Notebook
}
JupyterLab
You can also specify /usr/local/bin/jupyter-lab to jupyterCommand to utilize
Jupyter Lab (you may also want to install jupyterlab via pip3 install jupyterlab.)
pyenv
Put ~/.pyenv/shims/jupyter into jupyterCommand if you are on pyenv-enabled
environment.
Launch Juno from Terminal
Add juno command to open Jupyter notebooks from Terminal. Put following code
to your shell config file.
Applying for a place for your child at primary or secondary school in England can be a daunting task. In some regions, parents have a plethora of schools to choose from. Having options is no bad thing, but in order to make an informed choice parents need answers to questions such as:
How many preferences can I list on my application form?
What is the likelihood of getting an offer at my preferred school based on my circumstances?
Which schools are oversubscribed?
Which schools are the top performers in my area?
This dashboard sets out to help parents answer those questions and more in order to better understand the landscape before submitting their application. The dashboard uses data published by UK Government on school admissions and performance at both primary and secondary level, and covers all local authorities in England.
Contributions are what make the open source community such an amazing place to learn, inspire, and create. Any contributions you make are greatly appreciated.
If you have a suggestion that would make this better, please fork the repo and create a pull request. You can also simply open an issue with the tag “enhancement”.
Don’t forget to give the project a star! Thanks again!
Fork the Project
Create your Feature Branch (git checkout -b feature/AmazingFeature)
Commit your Changes (git commit -m 'Add some AmazingFeature')
Push to the Branch (git push origin feature/AmazingFeature)
The objective was only to see the behaviour of the training with a small amount of data. (Spot some mistakes and see the
behaviour of the loss)
To visualize our training performance we used the tool tensorboard. The default logdir in
in .cache/logs/$EXPERIMENT
where $EXPERIMENT is specified in --experiment. The log could be changed using flag --logdir or shorthand -s
This repository contains the python package for collecting, collating, manipulating, analyzing, and visualizing MRI data generated on brainlife.io. Designed to used within the brainlife.io Analysis tab Jupyter notebooks, can be installed as a pypi package to your local machine.
indelope: find indels and SVs too small for structural variant callers and too large for GATK
indelope was started with the goal of increasing the diagnostic rate in exomes. To do this it must be:
fast : ~2.5 CPU-minutes per exome (25% slower than samtools view -c)
easy-to-use : goes from BAM to VCF in a single command.
novel : it does local assembly and then aligns assembled contigs
to the genome to determine the event, and then does k-mer counting (not alignment) to genotype
without k-mer tables.
accurate : because of the genotyping method, we know that called variants are not present
in the reference.
These features will help ensure that it is actually used (fast, easy-to-use) and that it finds new
and valid variation.
As of November 2017, indelope is working — it finds large indels that are clearly valid by visual
inspection that are missed by GATK/freebayes/lumpy.
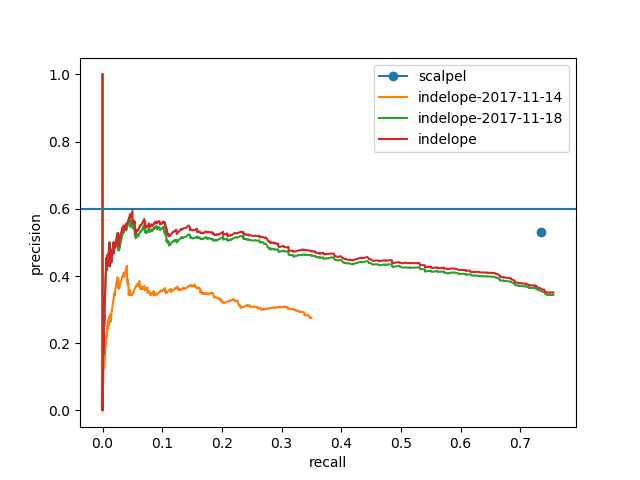
As of November 2017, I am still tuning. Here is a look at the progress:
Note that while indelope is steadily improving, it still is not as good as scalpel. More improvements
are coming soon.
indelope also works on whole genomes, but, for now, that is not the target use-case.
how it works
indelope sweeps over a single bam and finds regions that are likely to harbor indels–reads that have
more than 1 cigar event and split-reads (work on split reads is in progress). As it finds these it increments
a counter for the genomic position of the event. Upon finding a gap in coverage, it goes back, finds any
previous position with sufficient evidence (this is a parameter) of an event, gathers reads that have been
aligned across that genomic position (and unaligned reads from that region) and does assembly on those reads.
It then aligns the assembled contigs to the genome using ksw2 and uses the CIGAR
to determine the event as it’s represented in the VCF. Any event will result in a novel k-mer not present in
the reference genome; indelope gets the k-mer of the reference genome at the event and the novel k-mer of
the alternate event. It again iterates through the reads that were originally aligned to the event and counts
reference and alternate k-mers. Those counts are used for genotyping. Note that this reduces reference bias
because we are aligning a contig (often >400 bases) to the genome and never re-aligning the actual reads.
As indelope sweeps across the genome, it keeps the reads for each chunk in memory. A chunk bound is defined
by a gap in coverage; this occurs frequently enough that the memory use is negligible. Once a new chunk is reached,
all events from the previous chunk are called and then those reads are discarded. This method, along with the
assembly method make indelope extremely fast–given 2 BAM decompression threads, it can call variants in an
exome in ~ 1 minute (2.5 CPU-minutes).
assembly
A read (contig) slides along another read (contig) to find the offset with the most matches. At each offset, if
more than $n mismatches are found, the next offset is attempted. This is efficient enough that a random read to
a random (non-matching) contig of length $N will incur ~ 1.25 * $N equality (char vs. char) tests.
Within each contig indelope tracks the number of reads supporting each base. Given a sufficient number of
reads supporting a contig, it can account for sequencing errors with a simple voting scheme. That is: if contig a,
position x has more than 7 supporting reads and contig b has fewer than 3 supporting reads (and we know that
otherwise, a and b have no mismatches), we can vote to set the mismatch in b to the apparent match in a.
This allows us to first create contigs allowing no mismatches within the reads and then to combine and extend contigs
using this voting method.
installation and usage
get a binary from here
and make sure that libhts.so is in your LD_LIBRARY_PATH
somatic mode / filter mode. allow filtering on a set of k-mers from a parental genome (parent for
mendelian context or normal for somatic variation).
use SA tag. (and possibly discordant reads)
see also
svaba does local assembly, but then genotypes by alignment to those
assemblies. It is slower than indelope but it is an extremely useful tool and has a series of
careful and insightful analyses in its paper. (highly recommend!!)
rufus does k-mer based variant detection; Andrew described
to me the RUFUS assembly method that inspired the one now used in indelope.
lancet, scalpel,
mate-clever, and prosic2 are all
great tools that are similar in spirit that are worth checking out (of those, AFAICT, only scalpel has a focus on germ-line variation).
notes and TODO
need a better way to combine contigs
sometimes, can have 2 contigs, each of length ~ 80 and they overlap for 60 bases but cutoff is
e.g. 65. Need a way to recover this as it happens a lot in low-coverage scenarios. maybe it can
first combine, then trim (currently, it’s trim, combine).
This should also allow more permissive overlaps if the correction list is empty.
track a read/contig matches multiple contigs with the same match, mismatch count
min_overlap in contig::best_match should be a float between 0 and 1 that will make sure that at least
that portion of the shortest contig overlaps the other.





















{ "jupyterCommand": "/usr/local/bin/jupyter-notebook", // executable path for Jupyter Notebook "jupyterPort": 8888, // server port "jupyterHome": "~", // root folder "openBrowserOnStartup": true, // set true if let Juno open browser after launch "preferLab": false // open Jupyter Lab instead of Jupyter Notebook }